- Genome Sequencing on HiFi Sequencing
- Human Genome Sequencing for SV discovery
- ISO Sequencing
- Metagenome Sequencing on Sequel II
- Full length 16s Amplicon Sequencing on PacBio Sequel II
- Microbial Genome Sequencing
Nucleome Informatics Whole Genome Sequencing (WGS) Services with PacBio Revio HiFi Sequencing
Nucleome Informatics delivers state-of-the-art whole genome sequencing (WGS) servicesthrough the PacBio Revio system—the world’s most advanced long-read platform—now powered by SPRQ chemistry. We are the only company in India providing this technology, enabling researchers to achieve comprehensive and highly accurate genome assemblies for microbes, plants, and animals.
Key Features of Nucleome’s Revio WGS Services
-
HiFi Accuracy: Achieve single-molecule, long-read accuracy (≥99.9%, Q30+) across even the most complex genomes, capturing SNVs, indels, structural variants, tandem repeats, and methylation patterns.
-
Low DNA Input: Only 500 ng of high molecular weight, impurity-free DNA per sample is required—ideal for precious or limited samples.
-
Comprehensive Variant Detection: HiFi sequencing on Revio detects all variant types with phasing, enabling deeper insights into genetic diversity and functionality.
-
True Gapless Assemblies: Construct complete genomes, including repetitive and challenging regions like telomeres and centromeres, with long HiFi reads spanning tens of kilobases.
-
Uniform Coverage: SPRQ chemistry delivers even coverage across all genomic contexts, including high-GC and repetitive regions, without PCR-based biases.
-
Direct Methylation Detection: Analyze cytosine methylation and other modifications directly from native DNA—no additional library prep required.
-
Scalability: Handle projects from a few samples to thousands annually, enabling both individual projects and population-scale studies.
Sample Requirements
-
DNA Input: Minimum 500 ng of high molecular weight genomic DNA (ideally >20–30 Kb fragment size, >70% over 10 Kb for most applications).
-
Quality: DNA must be highly pure, with no contamination or degradation; quantification by Qubit or similar recommended.
-
Low Input Option: For extremely limited material, consult our technical team for specialized low-input workflows.
-
gDNA Extraction: If required, Nucleome offers DNA extraction and QC services from tissue or cell samples.
Quality Control and Library Preparation
-
QC Techniques: Qubit, Bioanalyzer, FEMTO Pulse, and PFGE for size distribution and purity assessment.
-
Shearing & Size Selection: Megaruptor 3 for fragmentation and BluePippin for size selection (eliminating sub-10 Kb fragments).
-
Library Construction: SMRTbell® library prep targeting 15–20 Kb insert sizes for de novo and variant detection projects.
Sequencing and Data Analysis
-
Coverage Guidelines: Tailored to project type and genome complexity—diploid genomes (30–50x), polyploid/haplotype-resolved assemblies (30x/haplotype), complex/repetitive genomes (up to 120x), microbial genomes (200x).
-
Comprehensive Analysis Pipeline:
-
De novo assembly (including mosaic and haplotype-phased)
-
Trio binning and microbial genome assembly
-
Polyploid assembly
-
Scaffolding with Optical Mapping and Hi-C data
-
Polishing, error correction, and curation
-
Variant detection (SNVs, indels, SVs, tandem repeats, and methylation)
-
Genome QC metrics: Contig N50/Scaffold N50, BUSCO score, RNA-Seq mappability, and chromosomal continuity
-
Applications
-
Human, animal, and plant whole genome sequencing
-
Microbial genomics (bacteria, fungi, archaea)
-
Structural and rare variant discovery
-
Epigenetic analysis (direct methylation calls)
-
Complex genome assembly—from crops to conservation species
Ready to unlock the fullest view of genetic diversity in your research project?
Contact Nucleome’s sales team at sales@nucleomeinfo.com for technical guidance or a tailored quote.
Choose Nucleome for highly accurate, comprehensive WGS powered by the PacBio Revio and HiFi sequencing advantage.


HiFi Sequencing Service for Structural Variant (SV) Discovery in Human Genomes
Unlock the full potential of genomic insights with Nucleome’s cutting-edge HiFi Sequencing service, designed to provide highly accurate and comprehensive structural variant (SV) detection in human genomes. Leveraging PacBio’s revolutionary HiFi reads, our service delivers unparalleled resolution for complex genetic analysis, making it an ideal choice for researchers studying rare and inherited diseases.
Why Choose HiFi Sequencing for SV Discovery?
HiFi reads combine long-read length with exceptional accuracy (99.9%), empowering researchers to:
- Detect all variant types, including single nucleotide variants (SNVs), indels, and structural variants (SVs) with best-in-class precision.
- Achieve complete and phased assemblies of the human genome, capturing regions inaccessible to other technologies, such as HLA and CYP2D6.
- Analyze multiomic profiles with simultaneous 5-methylcytosine (5mC) detection, eliminating the need for separate bisulfite sequencing workflows.
- Minimize errors and overcome challenges like GC bias and coverage gaps seen in traditional methods.
Structural Variant (SV) Detection
Variation between two human genomes, by the number of base pairs impacted, reveals that single-nucleotide variants (SNVs) are the most numerous variant type (~4–5 M/person), followed by indels. Structural variants (SVs) and tandem repeats, while fewer in number, account for more genomic variation between individuals due to their size (>50 bp). This makes them critical for understanding genetic diversity and disease associations.
HiFi sequencing stands out by offering accurate small variant calls alongside significantly more structural variant detection compared to other technologies. This comprehensive resolution enables researchers to:
- Study human genetic diversity in detail.
- Better elucidate function and disease association.
- Overcome the static solve rates of short-read whole genome sequencing (WGS) and whole exome sequencing (WES).
Key Features of Our HiFi Sequencing Service
- High Accuracy and Long Reads
- Per-base accuracy of 99.9%, ensuring reliable SV detection.
- Read lengths of 15-18 kb, perfect for resolving complex genomic regions.
- Comprehensive Variant Detection
- Identify SNVs, indels, and SVs with precision.
- Detect pathogenic repeat expansions and novel rearrangements.
- Integrated Epigenomic Analysis
- Simultaneous detection of 5mC profiles for a deeper understanding of disease mechanisms.
- Phased haplotypes for insights into allele-specific methylation.
- Scalable and Efficient Workflow
- High-throughput capability with up to 1300 and 256 samples sequenced per Revio and Sequel IIe systems annually.
- Automation-friendly library preparation to streamline processing of up to 96 libraries per week by a single technician.
- Data Analysis and Interpretation
- Open-source tools for alignment-based variant calling and de novo assembly.
- Expert visualization and interpretation to identify candidate variants and complex rearrangements.
Applications of HiFi Sequencing in Rare and Inherited Disease Research
HiFi sequencing is transforming how researchers approach rare disease diagnostics by:
- Identifying causative variants in cases unresolved by short-read sequencing.
- Discovering novel disease-associated genes through complete genome assemblies.
- Analyzing pathogenic repeat expansions, such as CTG triplet repeats in the DMPK gene associated with myotonic dystrophy.
- Enabling methylation profiling to study the epigenetic basis of disease.
HiFi Sequencing for Somatic Cancer Variant Discovery
The extraordinary accuracy and long read lengths of PacBio® HiFi sequencing overcome the challenges of detecting variation in cancer genomes faced by other technologies.
High-quality HiFi reads detect a wide range of cancer-specific somatic variation, including single nucleotide variants (SNVs), structural variants (SVs), insertions and deletions (indels), copy number variations (CNVs), and methylation in a single assay. With improved somatic variant calling accuracy, HiFi sequencing requires less sequencing relative to other technologies, making it an efficient solution for cancer research.
Advantages for Somatic Variant Discovery
- Accuracy: HiFi sequencing yields greater somatic variant calling accuracy with fewer sequencing reads compared to other platforms.
- Comprehensive Analysis: Detect SNVs, indels, SVs, and CNVs alongside methylation profiling for a holistic view of cancer genomes.
- Streamlined Workflow: From sample preparation to analysis, HiFi sequencing provides a cohesive workflow for tumor-normal whole genome sequencing, enhancing efficiency and scalability.
Workflow Overview
- Sample and Library Preparation
- Suitable for various sample types, including blood, saliva, tissue, and cultured cells.
- Optimized DNA shearing and library preparation for long-read sequencing.
- HiFi Sequencing
- Scalable sequencing capacity for large projects or smaller batches with reduced runtimes.
- Robust performance for high-accuracy variant detection.
- Data Analysis
- Comprehensive variant calling and de novo assembly using tools like pbmm2, DeepVariant, pbsv, and hifiasm.
- Visualization and interpretation of candidate variants via IGV and other platforms.
Partner with Nucleome for Advanced Genomic Insights
As an experienced long-read sequencing technology user, Nucleome Informatics is committed to providing world-class sequencing solutions. Our HiFi Sequencing service for SV discovery and somatic cancer variant detection empowers researchers to uncover the genetic basis of diseases with unmatched accuracy and efficiency.
Contact us today to explore how HiFi sequencing can revolutionize your research.
ISO Sequencing
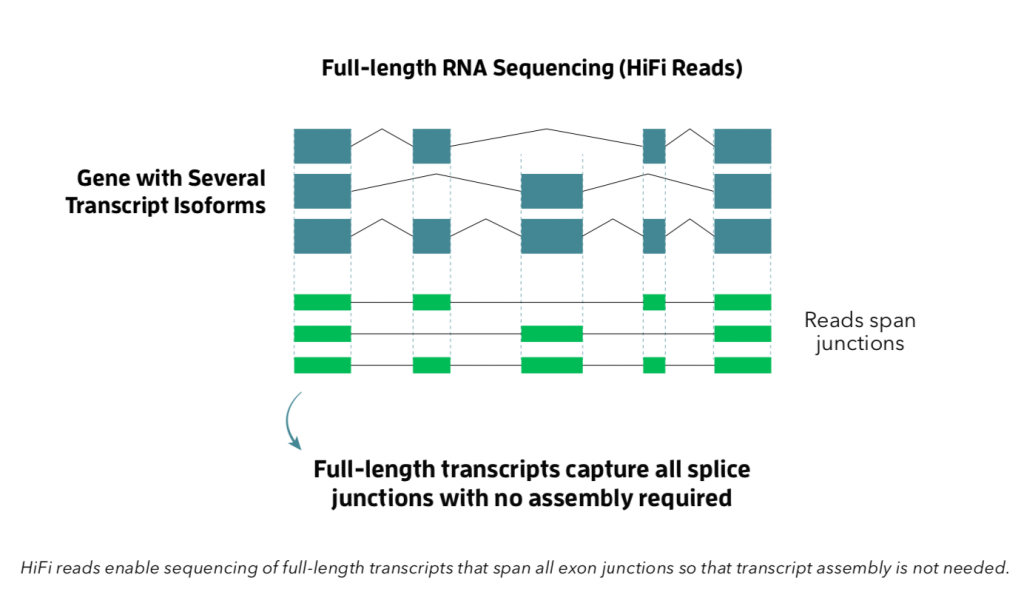
Nucleome provides full-length transcriptome sequencing i.e ISO Sequencing on Pacbio Sequel II platform. Nucleome Informatics is the only service provider of ISO Sequencing on Sequel II in India. The PacBio Iso-Seq method is an end-to-end workflow for sequencing and analyzing full-length transcript isoforms. Our Iso-Seq analysis enables you to detect novel transcripts and genes, identify fusion genes, annotate isoforms and alternative splicing events.
Generate full-length transcript sequences up to ~15kb
- High accuracy (>99%) for ORF prediction
- No reference genome required
- Bioinformatics tools from raw data to functional annotation
ISO Seq Service WorkFLow:
1. Convert RNA into cDNA
2. cDNA SMRTbell library preparation
3. Sequence on the Sequel II System
4. Generate circular consensus sequences (CCS)
5. Discover isoforms de novo with Iso-Seq analysis
ISO Seq Applications:
Whole-genome Annotation
-Typically whole-transcriptome, non- quantitative
-Often included in de novo genome assembly projects
-Single tissue to several tissues
-Generates reference transcriptome for downstream RNA-seq studies
Gene-level Isoform Discovery
-Typically targeted, either cDNA amplicons or target capture
-Useful for detecting gene fusions, SNVs, allele-specific expression
-Cost-effectively multiplex many samples per single SMRT Cell
-Relative quantitation possible
Sample Requirement
1 ug of high qulaity total RNA, RIN >7 and Concentration should be more than 25ng per ul
– Has not been exposed to high temperatures (e.g.: > 65°C for 1 hour can cause a detectable decrease in sequence quality) or pH extremes (< 6 or > 9).
– Has an OD260/OD280 ratio between 2.0 and 2.2. – Has an OD260/OD230 ratio between 1.8 and 2.1. – Has a RIN number ≥ 8 (Recommended).
– Has not been exposed to intercalating fluorescent dyes or ultraviolet radiation. SYBR dyes are not RNA damaging, but do avoid ethidium bromide.
– Does not contain denaturants (e.g., guanidinium salts or phenol) or detergents (e.g., SDS or Triton‐X100).
– Does not contain carryover contamination from the original organism/tissue (e.g., heme, humic acid, polyphenols, etc.).
– Note: RNA samples should only be shipped with dry ice.
Contact our team now to receive the quote and place the order.
Nucleome is the only service provider in South Asia that offers Single Molecule, Real-Time (SMRT) Sequencing on PacBio Sequel II that generates the long, accurate, single-molecule reads you need to comprehensively characterize samples with complex variation. With SMRT Sequencing you can:
- Identify closely related individuals within a heterogeneous mixture
- Track the rapid evolution in response to environmental conditions, immune pressures, or drug treatments
Highly accurate long reads – HiFi reads – with the single-molecule resolution are ideal for full-length 16S rRNA sequencing, shotgun metagenomic profiling, and metagenome assembly so that you can:
- Determine community composition at the species or strain-level with competitively priced full-length16S sequencing
- Identify 6-8 full-length genes in every HiFi read with efficient, cost-effective metagenomic profiling
- Generate up to 20 high-quality metagenome-assembled genomes (MAGs) per multiplexed human faecal sample with just 6 Gb of HiFi data
- Leverage epigenomic data to associate contigs and plasmids from closely related strains
Full-length Metagenome assembly and functional profiling on PacBio Sequel II
HiFi sequencing on the PacBio Sequel II System enables complete microbial community profiling of complex metagenomic samples using whole genome shotgun sequences. With HiFi sequencing, highly accurate long reads overcome the challenges posed by the presence of intergenic and extragenic repeat elements in microbial genomes, thus greatly improving phylogenetic profiling and sequence assembly.
HiFi sequencing can be used even for sample types with limited starting material, allowing for reference quality MAGs, discovery of novel genes without the need for de novo assembly, identification of biochemical pathways, and resolution of taxa below the species level with confidence.
- Unbiased compositional and functional characterization of microbial communities
- Generation of complete or near- complete genome assemblies from microbial populations
Data analysis:
Output data in standard file formats, (BAM and FASTA/Q) for seamless integration with downstream analysis tools – Perform taxonomic classification and functional gene profiling using QIIME and MEGAN
– Perform gene prediction and discovery using FragGeneScan and Prodigal
Learn more about workflows for these types of complex populations
- Resolve viral populations – to understand evolution, quasispecies dynamics, drug-resistance, and immune escape
- Characterize microbial communities – with the species-level resolution with full-length 16S rDNA and shotgun sequencing for metagenomic profiling and assemblies
- Detect somatic variation – to explore compound mutations and splice isoforms in cancer cell populations
– Perform metagenomic shotgun assembly directly with HiFi reads using HiCanu

Full length 16s Amplicon Sequencing by Nucleome
Nucleome is the only service provider in South Asia that offers Single Molecule, Real-Time (SMRT) Sequencing on PacBio Sequel II that generates full length 16s amplicon sequencing. The ~1500 bp 16S rRNA gene comprises nine variable regions interspersed throughout the highly conserved 16S sequence. Sequencing the partial gene was originally accomplished by Sanger sequencing. This required cloning genes, generating, and assembling two to three reads per clone, and producing limited sampling depth at high cost and effort. The partial 16s rRNA gene sequencing misses to sequence the entire taxa of the bacterial community.
DIFFERENT SUB-REGIONS SHOWED BIAS IN THE BACTERIAL TAXA THEY WERE ABLE TO IDENTIFY
- V4: Consistently poor performance
- V1–V2: poor for Proteobacteria
- V3–V5: poor for Actinobacteria
- V1–V3: good results for Escherichia / Shigella
- V3–V5: good results for Klebsiella,
- V6–V9: good results for Clostridium and Staphylococcus
Full V1–V9 region: the only way to resolve ALL the clades that may be present in a complex population. Order full length 16s Amplicon sequencing to Nucleome and discover Strain-level identification of bacterial communities with the unprecedented accuracy of PacBio HiFi reads.

Microbial Genomics Service on Pacbio Sequel II
Nucleome Informatics embraces the power of PacBio Sequel II’s HiFi sequencing technology to shed light on the intricate world of microbial genomes. With this cutting-edge platform, we’ve unlocked the potential to decipher the genetic codes of bacteria, viruses, bacteriophages, and fungal organisms like never before. In this article, we’ll explore how HiFi sequencing on the PacBio Sequel II platform is redefining our understanding of these microscopic life forms. Microbial genome sequencing has revolutionized our understanding of the microbial world. PacBio Sequel II, with its cutting-edge technology, has emerged as a game-changer in the field of microbial genomics. This state-of-the-art platform offers unparalleled advantages for sequencing microbial genomes, enabling researchers to delve deeper into the genetic diversity and functional potential of microorganisms. In this article, we will explore how PacBio Sequel II empowers scientists to unravel the mysteries of microbial genomes.
The Power of Long Reads:
One of the key features that sets PacBio Sequel II apart is its ability to produce long-read sequences. Traditional short-read sequencing technologies often struggle with complex microbial genomes due to their fragmented assembly. PacBio’s Single Molecule, Real-Time (SMRT) sequencing technology generates long reads, making it easier to assemble and analyze microbial genomes, especially those with high GC content or repetitive regions.
High Accuracy and Low Error Rates:
PacBio Sequel II boasts impressive accuracy levels, even in regions that are challenging for other sequencing platforms. With the use of circular consensus sequencing (CCS), it is possible to obtain highly accurate HiFi reads (>99.99% accuracy). This is crucial when studying microbial genomes, as even a single base-pair error can significantly impact downstream analysis, such as functional gene prediction or comparative genomics.
Unraveling Genomic Complexity:
Microbial genomes are incredibly diverse and can contain complex structures, such as plasmids, transposons, and repeat elements. PacBio Sequel II’s long reads and high accuracy help researchers decipher these intricate genomic landscapes with ease. This technology is particularly valuable for investigating microbial populations, identifying genomic variations, and understanding horizontal gene transfer events.

Resolution of Epigenetic Modifications:
In addition to the genetic code, epigenetic modifications play a critical role in microbial function. PacBio Sequel II can identify and characterize DNA modifications, such as methylation patterns, providing insights into gene regulation, adaptation, and virulence mechanisms within microbial populations.
Applications in Microbial Genomics:
The versatility of PacBio Sequel II extends to a wide range of applications in microbial genomics:
- Microbial Diversity Studies: Explore the richness and diversity of microbial communities in various environments, from soil to the human microbiome.
- Functional Genomics: Understand the functional potential of microbes by annotating complete genomes and identifying important genes and pathways.
- Antimicrobial Resistance: Investigate the genetic basis of antibiotic resistance in pathogens, aiding in the development of targeted treatments.
- Phylogenetic Analysis: Build accurate phylogenetic trees and trace the evolutionary history of microorganisms.
- Metagenomics: Analyze complex microbial ecosystems without the need for culturing individual strains.
Contact us now for receiving the proposal for microbial genome sequencing.